Scleroderma
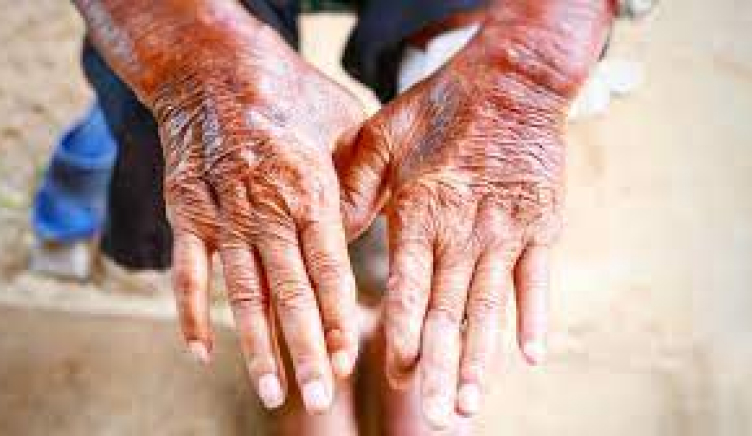
Scleroderma (including systemic sclerosis, SSc): a rare and potentially life-threatening autoimmune disorder characterised by inflammation (thickening) and fibrosis (hardening/scarring) of the skin and other organs. Scleroderma results in high morbidity and has the highest mortality among rheumatic diseases.
The inflammation and fibrosis in scleroderma affect systemic connective tissue. Characteristics of scleroderma include essential vasomotor disturbances; fibrosis; subsequent atrophy of the skin, subcutaneous tissue, muscles, and internal organs (e.g., alimentary tract, lungs, heart, kidney, CNS); and immunologic disturbances accompany these findings.
Scleroderma is a progressive disease, with substantial detriment on the quality of life for patients, who experience loss of mobility and function, pain, fatigue, often with a significant impact to their mental health.
There are different prevalence estimates for scleroderma among different world populations, with higher population estimates in the US than in Europe or Asia; overall this is a rare (orphan) disease. Latest data suggests that the prevalence of scleroderma is approximately 250 per million adults in the US (80-100,000 patients), 233 per million in Australia (~6,000 patients), 88-158 per million in Western Europe.
Women are affected much more frequently than men, with a female-to-male ratio of about 8:1 and most commonly presents between the ages of 30-40 years.
The prognosis depends on the type of scleroderma and the extent of organ involvement in the disease. Severe cases where the inflammation and fibrosis occur in multiple organs results in a high mortality rate without rapid intervention. Survival complicated by pulmonary hypertension remains poor despite currently available treatment options; other major organ involvement resulting in poor outcomes include kidneys and the heart.
Treatment goals are based on the distribution of organ involvement. Treatment is targeted on disease processes that are potentially reversible (e.g., active inflammation or vasoconstriction) and aims to minimise the patient’s functional impairment. It is common to describe the treatments for scleroderma according to system involvement. However, as scleroderma may be manifested in a variety of bodily systems, individual patients may require multi-modal therapies.
Standard of care relies on immunosuppression therapy and biologic agents which are not well tolerated. Severe and life-threatening renal disease develops in 10% to 15% of scleroderma patients. However, pharmacologic options for these patients are limited to treatment of hypertension with angiotensin-converting enzyme (ACE) inhibitors.
There is a significant medical need for a safe, well tolerated and efficacious anti-fibrotic therapy for scleroderma, which may reduce inflammation and fibrotic disease progression, and offer quality of life benefits for patients.
Disclaimer
All of the candidate drugs described on this site are investigational products which have not received marketing authorization or approval by any regulatory agency, including the US Food and Drug Administration, the European Medicines Agency, or the Australian Therapeutic Goods Agency. The investigational drug products are undergoing clinical studies to evaluate the safety and effectiveness in humans.